Reblozyl
What is Reblozyl (Luspatercept)?
Approved To Treat
Top Local Experts
There are no experts for this drug
Related Clinical Trials
Summary: This is a Phase 2a study to evaluate the safety and pharmacokinetics (PK) of luspatercept in pediatric participants with β-thalassemia. The study will be conducted in 2 parts for both transfusion-dependent (TD) and non-transfusion-dependent (NTD) β-thalassemia participants: TD Part A will be in adolescent participants aged 12 to \<18 years with two dose escalation cohorts, followed by a dose expan...
Summary: The purpose of the study is to compare the efficacy and safety of Luspatercept vs epoetin alfa in the treatment of anemia in adults due to IPSS-R very low, low, intermediate-risk MDS in ESA-naïve participants who are non-transfusion dependent (NTD).
Summary: The purpose of the study is to see if participants with anemia due to their type of MDS or MDS/MPN will experience a more decreased need for regular blood transfusions if they take luspatercept plus best supportive care, and what effect, good and/or bad, luspatercept has on them and their anemia due to MDS or MDS/MPN. The safety and tolerability of luspatercept will also be evaluated in this study...
Related Latest Advances
Brand Information
- For injection: 25 mg white to off-white lyophilized powder in a single-dose vial for reconstitution.
- For injection: 75 mg white to off-white lyophilized powder in a single-dose vial for reconstitution.
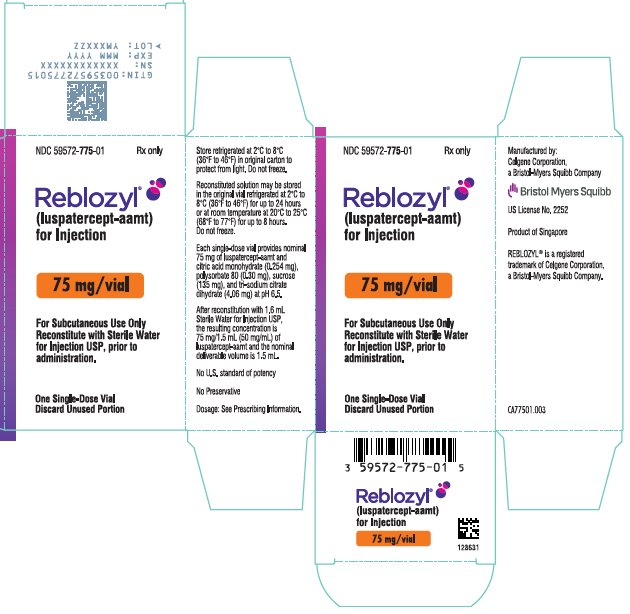
REBLOZYL 75 mg/vial (NDC 59572-775-01)
(luspatercept-aamt)
for Injection
Reconstitute prior
to administration
EXP

Rx only
(luspatercept-aamt)
for Injection
Reconstitute with Sterile Water
for Injection USP, prior to
administration.
Discard Unused Portion
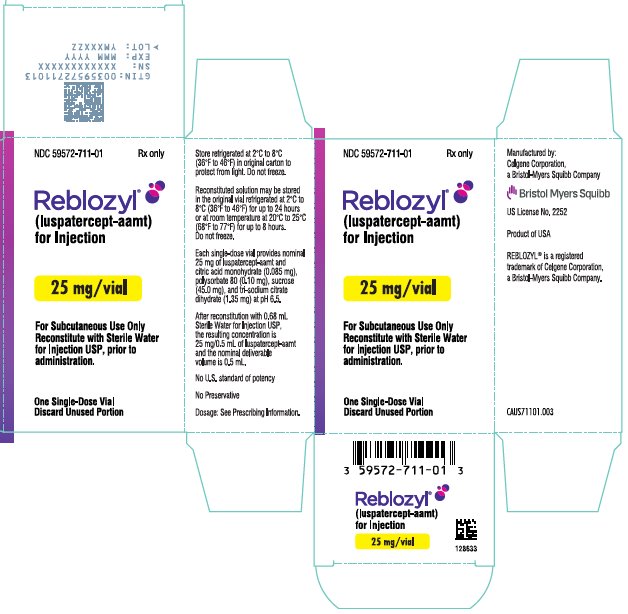
Rx only
(luspatercept-aamt)
for Injection
Reconstitute with Sterile Water
for Injection USP, prior to
administration.
Discard Unused Portion
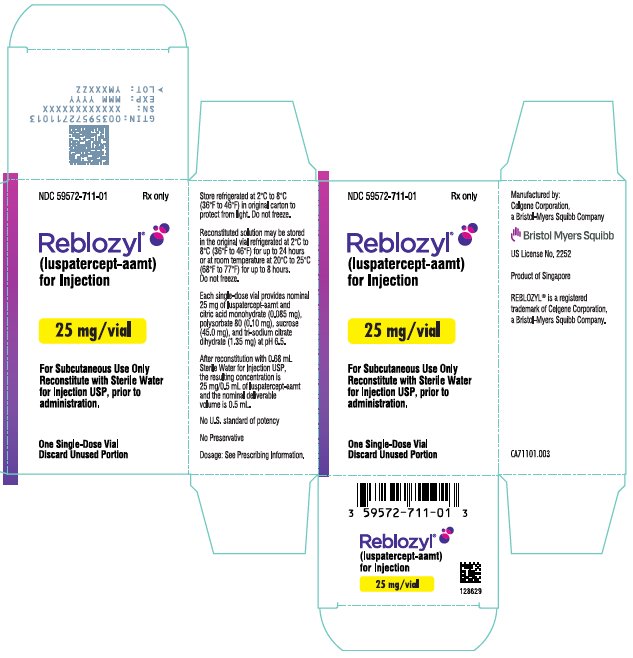
(luspatercept-aamt)
for Injection
Reconstitute prior
to administration
EXP

Rx only
(luspatercept-aamt)
for Injection
Reconstitute with Sterile Water
for Injection USP, prior to
administration.
Discard Unused Portion
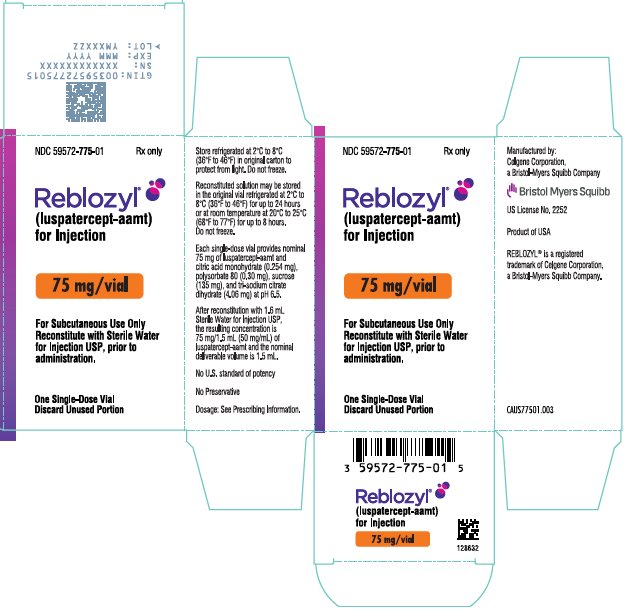
Rx only
(luspatercept-aamt)
for Injection
Reconstitute with Sterile Water
for Injection USP, prior to
administration.
Discard Unused Portion