Sutent
What is Sutent (Sunitinib)?
Top Local Experts
There are no experts for this drug
Related Clinical Trials
Summary: Main Objective of this study is to examine long-term safety of nivolumab monotherapy including combinations and other cancer therapies in various tumor types.
Summary: This study is a Phase 1/1b clinical trial that aims to determine the Maximally Tolerated Dose of Losartan and Sunitinib Combination Therapy. Patients will first be accrued to the Dose Escalation phase of the study, using a 3+3 design. Medication dosages will increase until a maximally tolerated dose is found. Patients will then be accrued to the Dose Expansion phase of the trial, where efficacy of...
Summary: This phase I trial tests the safety, side effects, and best dose of sunitinib malate in combination with lutetium Lu 177 dotatate in treating patients with pancreatic neuroendocrine tumors. Sunitinib malate is in a class of medications called kinase inhibitors and a form of targeted therapy that blocks the action of abnormal proteins called VEGFRs that signal tumor cells to multiply. This helps st...
Related Latest Advances
Brand Information
- 12.5 mg sunitinib: orange cap and orange body, printed with white ink "Pfizer" on the cap and "STN 12.5 mg" on the body.
- 25 mg sunitinib: caramel cap and orange body, printed with white ink "Pfizer" on the cap and "STN 25 mg" on the body.
- 37.5 mg sunitinib: yellow cap and yellow body, printed with black ink "Pfizer" on the cap and "STN 37.5 mg" on the body.
- 50 mg sunitinib: caramel top and caramel body, printed with white ink "Pfizer" on the cap and "STN 50 mg" on the body.
- Hepatotoxicity [see Warnings and Precautions (5.1)]
- Cardiovascular Events [see Warnings and Precautions (5.2)]
- QT Interval Prolongation and Torsade de Pointes [see Warnings and Precautions (5.3)]
- Hypertension [see Warnings and Precautions (5.4)]
- Hemorrhagic Events [see Warnings and Precautions (5.5)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.6)]
- Thrombotic Microangiopathy [see Warnings and Precautions (5.7)]
- Proteinuria [see Warnings and Precautions (5.8)]
- Dermatologic Toxicities [see Warnings and Precautions (5.9)]
- Reversible Posterior Leukoencephalopathy Syndrome [see Warnings and Precautions (5.10)]
- Thyroid Dysfunction [see Warnings and Precautions (5.11)]
- Hypoglycemia [see Warnings and Precautions (5.12)]
- Osteonecrosis of the Jaw [see Warnings and Precautions (5.13)]
- Impaired Wound Healing [see Warnings and Precautions (5.14)]
- Blood and lymphatic system disorders: hemorrhage associated with thrombocytopenia
including some fatalities . - Gastrointestinal disorders: esophagitis.
- Hepatobiliary disorders: cholecystitis, particularly acalculous cholecystitis.
- Immune system disorders: hypersensitivity reactions, including angioedema.
- Infections and infestations: serious infection (with or without neutropenia)
. The infections most commonly observed with SUTENT include respiratory, urinary tract, skin infections, and sepsis/septic shock. - Musculoskeletal and connective tissue disorders: fistula formation, sometimes associated with tumor necrosis and/or regression
; myopathy and/or rhabdomyolysis with or without acute renal failure . - Renal and urinary disorders: renal impairment and/or failure
. - Respiratory disorders: pulmonary embolism
, pleural effusion . - Skin and subcutaneous tissue disorders: pyoderma gangrenosum, including positive de-challenges.
- Vascular disorders: arterial (including aortic) aneurysms, dissections
, and rupture ; arterial thromboembolic events . The most frequent events included cerebrovascular accident, transient ischemic attack, and cerebral infarction. - General disorders and administration site conditions: impaired wound healing.
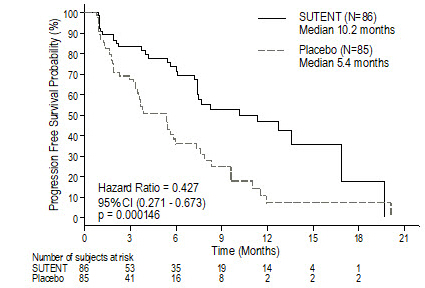
(sunitinib malate)
Rx only

(sunitinib malate)
Rx only

(sunitinib malate)
Rx only

(sunitinib malate)
Rx only
