Generic Name
Pembrolizumab
Brand Names
Keytruda, Keytruda QLEX
FDA approval date: January 15, 2015
Classification: Endoglycosidase
Form: Injection
What is Keytruda (Pembrolizumab)?
KEYTRUDA QLEX is a combination of pembrolizumab, a programmed death receptor-1 -blocking antibody, and berahyaluronidase alfa, an endoglycosidase, indicated: Melanoma for the treatment of adult patients with unresectable or metastatic melanoma.
Approved To Treat
Top Global Experts
There are no experts for this drug
Save this treatment for later
Not sure about your diagnosis?
Tired of the same old research?
Related Clinical Trials
There is no clinical trials being done for this treatment
Related Latest Advances
There is no latest advances for this treatment
Brand Information
KEYTRUDA (pembrolizumab)
1DOSAGE FORMS AND STRENGTHS
- Injection: 100 mg/4 mL (25 mg/mL) clear to slightly opalescent, colorless to slightly yellow solution in a single-dose vial
2CONTRAINDICATIONS
None.
3ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling.
- Severe and fatal immune-mediated adverse reactions
- Infusion-related reactions
3.1Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS AND PRECAUTIONS reflect exposure to KEYTRUDA as a single agent in 2799 patients in three randomized, open-label, active-controlled trials (KEYNOTE-002, KEYNOTE-006, and KEYNOTE-010), which enrolled 912 patients with melanoma and 682 patients with NSCLC, and one single-arm trial (KEYNOTE-001), which enrolled 655 patients with melanoma and 550 patients with NSCLC. In addition to the 2799 patients, certain subsections in the WARNINGS AND PRECAUTIONS describe adverse reactions observed with exposure to KEYTRUDA as a single agent in a randomized, placebo-controlled trial (KEYNOTE-091), which enrolled 580 patients with resected NSCLC, a non-randomized, open-label, multi-cohort trial (KEYNOTE-012), a non-randomized, open-label, single-cohort trial (KEYNOTE-055), and two randomized, open-label, active-controlled trials (KEYNOTE-040 and KEYNOTE-048 single agent arms), which enrolled 909 patients with HNSCC; in two non-randomized, open-label trials (KEYNOTE-013 and KEYNOTE-087) and one randomized, open-label, active-controlled trial (KEYNOTE-204), which enrolled 389 patients with cHL; in a randomized, open-label, active-controlled trial (KEYNOTE-048 combination arm), which enrolled 276 patients with HNSCC; in combination with axitinib in a randomized, active-controlled trial (KEYNOTE-426), which enrolled 429 patients with RCC; and in post-marketing use. Across all trials, KEYTRUDA was administered at doses of 2 mg/kg intravenously every 3 weeks, 10 mg/kg intravenously every 2 weeks, 10 mg/kg intravenously every 3 weeks, or 200 mg intravenously every 3 weeks. Among the 2799 patients, 41% were exposed for 6 months or more and 21% were exposed for 12 months or more.
3.2Postmarketing Experience
The following adverse reactions have been identified during postapproval use of KEYTRUDA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal: Exocrine pancreatic insufficiency
Hepatobiliary: sclerosing cholangitis
4DESCRIPTION
Pembrolizumab is a programmed death receptor-1 (PD 1)-blocking antibody. Pembrolizumab is a humanized monoclonal IgG4 kappa antibody with an approximate molecular weight of 149 kDa. Pembrolizumab is produced in recombinant Chinese hamster ovary (CHO) cells.
KEYTRUDA (pembrolizumab) injection is a sterile, preservative-free, clear to slightly opalescent, colorless to slightly yellow solution for intravenous use. Each vial contains 100 mg of pembrolizumab in 4 mL of solution. Each 1 mL of solution contains 25 mg of pembrolizumab and is formulated in: L-histidine (1.55 mg), polysorbate 80 (0.2 mg), sucrose (70 mg), and Water for Injection, USP.
5HOW SUPPLIED/STORAGE AND HANDLING
KEYTRUDA injection (clear to slightly opalescent, colorless to slightly yellow solution):
Carton containing one 100 mg/4 mL (25 mg/mL), single-dose vial (NDC 0006-3026-02)
6PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
7PRINCIPAL DISPLAY PANEL - 50 mg Vial Carton
NDC 0006-3029-02
Keytruda®
(pembrolizumab)
(pembrolizumab)
50 mg /vial
For Intravenous Infusion Only
Dispense the enclosed Medication Guide to each patient.
Sterile lyophilized powder must be reconstituted with Sterile Water for
Rx only
Single-dose vial. Discard unused portion.

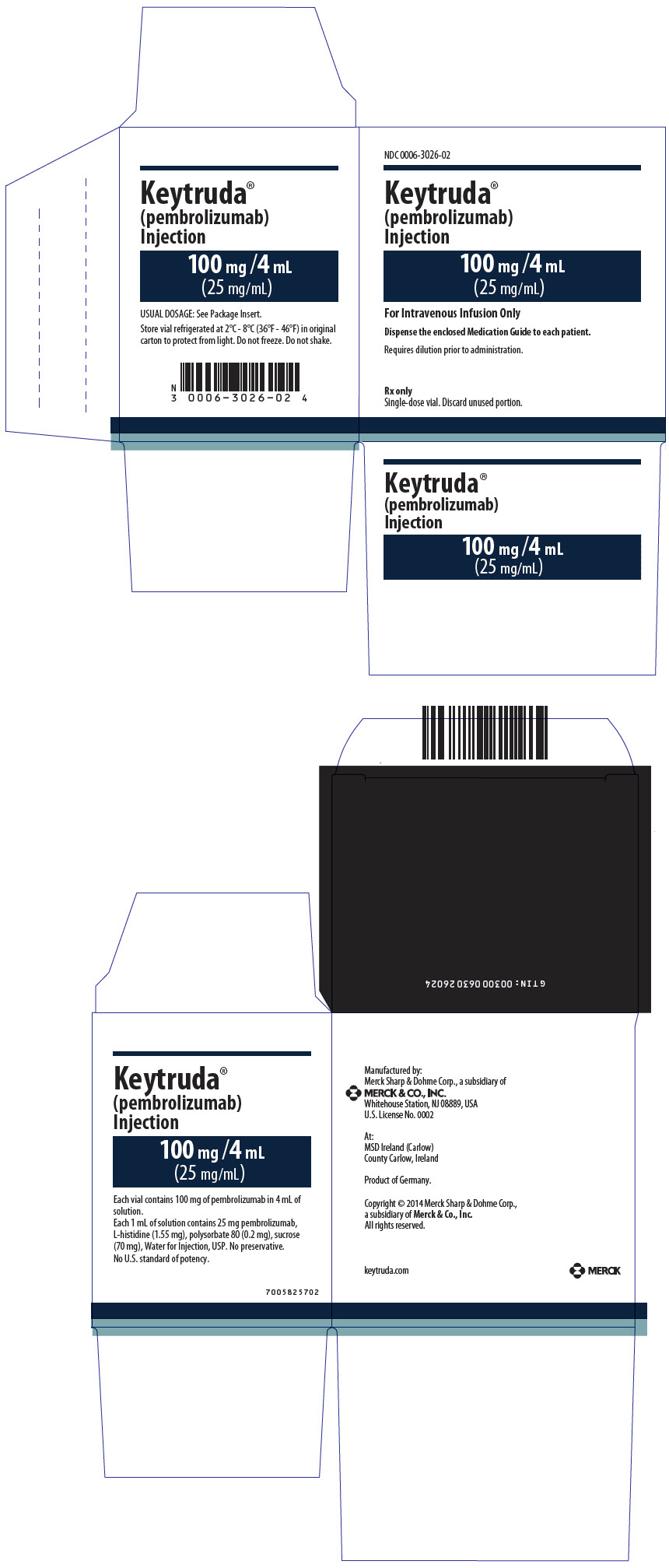
8PRINCIPAL DISPLAY PANEL - 100 mg/4 mL Vial Carton
NDC 0006-3026-02
Keytruda
100 mg /4 mL
(25 mg/mL)
(25 mg/mL)
For Intravenous Infusion Only
Dispense the enclosed Medication Guide to each patient.
Requires dilution prior to administration.
Rx only
Single-dose vial. Discard unused portion.
Single-dose vial. Discard unused portion.