Monoclonal antibodies directed against aggregated forms of beta amyloid, including LEQEMBI, can cause amyloid related imaging abnormalities (ARIA), characterized as ARIA with edema (ARIA-E), which can be observed on MRI as brain edema or sulcal effusions, and ARIA with hemosiderin deposition (ARIA-H), which includes microhemorrhage and superficial siderosis. ARIA can occur spontaneously in patients with Alzheimer’s disease, particularly in patients with MRI findings suggestive of cerebral amyloid angiopathy, such as pretreatment microhemorrhage or superficial siderosis. ARIA-H associated with monoclonal antibodies directed against aggregated forms of beta amyloid generally occurs in association with an occurrence of ARIA-E. ARIA-H of any cause and ARIA-E can occur together.
ARIA usually occurs early in treatment and is usually asymptomatic, although serious and life-threatening events, including seizure and status epilepticus, can occur. ARIA can be fatal. When present, reported symptoms associated with ARIA may include headache, confusion, visual changes, dizziness, nausea, and gait difficulty. Focal neurologic deficits may also occur. Symptoms associated with ARIA usually resolve over time. In addition to ARIA, intracerebral hemorrhages greater than 1 cm in diameter have occurred in patients treated with LEQEMBI.
Consider the benefit of LEQEMBI for the treatment of Alzheimer’s disease and potential risk of serious adverse events associated with ARIA when deciding to initiate treatment with LEQEMBI.
Incidence of ARIA
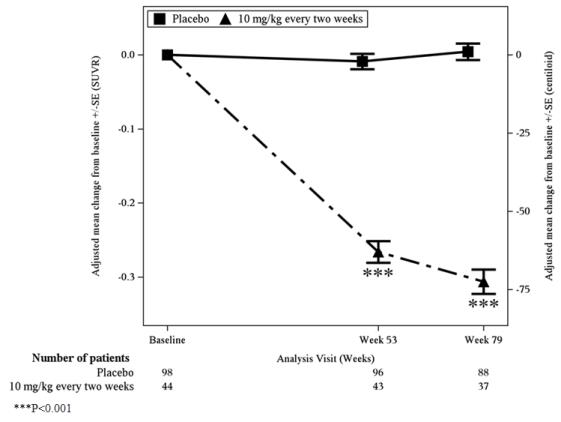
Symptomatic ARIA occurred in 3% (29/898) of patients treated with LEQEMBI in Study 2 [see Clinical Studies (14)]. Serious symptoms associated with ARIA were reported in 0.7% (6/898) of patients treated with LEQEMBI. Clinical symptoms associated with ARIA resolved in 79% (23/29) of patients during the period of observation. Similar findings were observed in Study 1. Including asymptomatic radiographic events, ARIA was observed in 21% (191/898) of patients treated with LEQEMBI, compared to 9% (84/897) of patients on placebo in Study 2.
In Study 2, ARIA-E was observed in 13% (113/898) of patients treated with LEQEMBI, compared to 2% (15/897) of patients on placebo. ARIA-H was observed in 17% (152/898) of patients treated with LEQEMBI, compared to 9% (80/897) of patients on placebo. There was no increase in isolated ARIA-H (i.e., ARIA-H in patients who did not also experience ARIA-E) for LEQEMBI compared to placebo.
Incidence of Intracerebral Hemorrhage
Intracerebral hemorrhage greater than 1 cm in diameter was reported in 0.7% (6/898) of patients in Study 2 after treatment with LEQEMBI, compared to 0.1% (1/897) on placebo. Fatal events of intracerebral hemorrhage in patients taking LEQEMBI have been observed.
Risk Factors for ARIA and Intracerebral Hemorrhage
ApoE ε4 Carrier Status
The risk of ARIA, including symptomatic and serious ARIA, is increased in apolipoprotein E ε4 (ApoE ε4) homozygotes.
Approximately 15% of Alzheimer’s disease patients are ApoE ε4 homozygotes. In Study 2, 16% (141/898) of patients in the LEQEMBI arm were ApoE ε4 homozygotes, 53% (479/898) were heterozygotes, and 31% (278/898) were noncarriers. The incidence of ARIA was higher in ApoE ε4 homozygotes (45% on LEQEMBI vs. 22% on placebo) than in heterozygotes (19% on LEQEMBI vs 9% on placebo) and noncarriers (13% on LEQEMBI vs 4% on placebo). Among patients treated with LEQEMBI, symptomatic ARIA-E occurred in 9% of ApoE ε4 homozygotes compared to 2% of heterozygotes and 1% noncarriers. Serious events of ARIA occurred in 3% of ApoE ε4 homozygotes, and approximately 1% of heterozygotes and noncarriers. The recommendations on management of ARIA do not differ between ApoE ε4 carriers and noncarriers [see Dosage and Administration (2.3)]. Testing for ApoE ε4 status should be performed prior to initiation of treatment to inform the risk of developing ARIA. Prior to testing, prescribers should discuss with patients the risk of ARIA across genotypes and the implications of genetic testing results. Prescribers should inform patients that if genotype testing is not performed, they can still be treated with LEQEMBI; however, it cannot be determined if they are ApoE ε4 homozygotes and at higher risk for ARIA. An FDA-authorized test for the detection of ApoE ε4 alleles to identify patients at risk of ARIA if treated with LEQEMBI is not currently available. Currently available tests used to identify ApoE ε4 alleles may vary in accuracy and design.
Radiographic Findings of Cerebral Amyloid Angiopathy (CAA)
Neuroimaging findings that may indicate CAA include evidence of prior intracerebral hemorrhage, cerebral microhemorrhage, and cortical superficial siderosis. CAA has an increased risk for intracerebral hemorrhage. The presence of an ApoE ε4 allele is also associated with cerebral amyloid angiopathy.
The baseline presence of at least 2 microhemorrhages or the presence of at least 1 area of superficial siderosis on MRI, which may be suggestive of CAA, have been identified as risk factors for ARIA. Patients were excluded from enrollment in Study 2 for the presence of more than 4 microhemorrhages and additional findings suggestive of cerebral amyloid angiopathy (prior cerebral hemorrhage greater than 1 cm in greatest diameter, superficial siderosis, vasogenic edema) or other lesions (aneurysm, vascular malformation) that could potentially increase the risk of intracerebral hemorrhage.
Concomitant Antithrombotic or Thrombolytic Medication
In Study 2, baseline use of antithrombotic medication (aspirin, other antiplatelets, or anticoagulants) was allowed if the patient was on a stable dose. The majority of exposures to antithrombotic medications were to aspirin. Antithrombotic medications did not increase the risk of ARIA with LEQEMBI. The incidence of intracerebral hemorrhage was 0.9% (3/328 patients) in patients taking LEQEMBI with a concomitant antithrombotic medication at the time of the event, compared to 0.6% (3/545 patients) in those who did not receive an antithrombotic. Patients taking LEQEMBI with an anticoagulant alone or combined with an antiplatelet medication or aspirin had an incidence of intracerebral hemorrhage of 2.5% (2/79 patients), compared to none in patients who received placebo.
Fatal cerebral hemorrhage has occurred in a patient taking an anti-amyloid monoclonal antibody in the setting of focal neurologic symptoms of ARIA and the use of a thrombolytic agent.
Additional caution should be exercised when considering the administration of antithrombotics or a thrombolytic agent (e.g., tissue plasminogen activator) to a patient already being treated with LEQEMBI. Because ARIA-E can cause focal neurologic deficits that can mimic an ischemic stroke, treating clinicians should consider whether such symptoms could be due to ARIA-E before giving thrombolytic therapy in a patient being treated with LEQEMBI.
Caution should be exercised when considering the use of LEQEMBI in patients with factors that indicate an increased risk for intracerebral hemorrhage and in particular for patients who need to be on anticoagulant therapy, or patients with findings on MRI that are suggestive of cerebral amyloid angiopathy.
Radiographic Severity
The radiographic severity of ARIA associated with LEQEMBI was classified by the criteria shown in Table 3.
In Study 2, the majority of ARIA-E radiographic events occurred early in treatment (within the first 7 doses), although ARIA can occur at any time and patients can have more than 1 episode. The maximum radiographic severity of ARIA-E in patients treated with LEQEMBI was mild in 4% (37/898) of patients, moderate in 7% (66/898) of patients, and severe in 1% (9/898) of patients. Resolution on MRI occurred in 52% of ARIA-E patients by 12 weeks, 81% by 17 weeks, and 100% overall after detection. The maximum radiographic severity of ARIA-H microhemorrhage in patients treated with LEQEMBI was mild in 9% (79/898), moderate in 2% (19/898), and severe in 3% (28/898) of patients; superficial siderosis was mild in 4% (38/898), moderate in 1% (8/898), and severe in 0.4% (4/898). Among patients treated with LEQEMBI, the rate of severe radiographic ARIA-E was highest in ApoE ε4 homozygotes 5% (7/141), compared to heterozygotes 0.4% (2/479) or noncarriers 0% (0/278). Among patients treated with LEQEMBI, the rate of severe radiographic ARIA-H was highest in ApoE ε4 homozygotes 13.5% (19/141), compared to heterozygotes 2.1% (10/479) or noncarriers 1.1% (3/278).
Monitoring and Dose Management Guidelines
Recommendations for dosing in patients with ARIA-E depend on clinical symptoms and radiographic severity [see Dosage and Administration (2.3)]. Recommendations for dosing in patients with ARIA-H depend on the type of ARIA-H and radiographic severity [see Dosage and Administration (2.3)]. Use clinical judgment in considering whether to continue dosing in patients with recurrent ARIA-E. Baseline brain MRI and periodic monitoring with MRI are recommended [see Dosage and Administration (2.3)]. Enhanced clinical vigilance for ARIA is recommended during the first 14 weeks of treatment with LEQEMBI. If a patient experiences symptoms suggestive of ARIA, clinical evaluation should be performed, including MRI if indicated. If ARIA is observed on MRI, careful clinical evaluation should be performed prior to continuing treatment. There is no experience in patients who continued dosing through symptomatic ARIA-E, or through asymptomatic but radiographically severe ARIA-E. There is limited experience in patients who continued dosing through asymptomatic but radiographically mild to moderate ARIA-E. There are limited data in dosing patients who experienced recurrent ARIA-E.
Providers should encourage patients to participate in real world data collection (e.g., registries) to help further the understanding of Alzheimer’s disease and the impact of Alzheimer’s disease treatments. Providers and patients can contact Eisai at 888-274-2378 for a list of currently enrolling programs.